Tumor Heterogeneity Analysis (THetA) is an algorithm used to estimate tumor purity and clonal/subclonal copy number aberrations simultaneously from high-throughput DNA sequencing data.
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --mem=14g --cpus-per-task=6
salloc.exe: Pending job allocation 46116226
salloc.exe: job 46116226 queued and waiting for resources
salloc.exe: job 46116226 has been allocated resources
salloc.exe: Granted job allocation 46116226
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn3144 are ready for job
[user@cn3144 ~]$ module load theta/0.7-20-g94fd772
[user@cn3144 ~]$ cp -r ${THETA_TEST_DATA:-none}/example/ .
[user@cn3144 ~]$ RunTHetA example/Example.intervals \
--NUM_PROCESSES=$((SLURM_CPUS_PER_TASK - 1)) \
--TUMOR_FILE example/TUMOR_SNP.formatted.txt \
--NORMAL_FILE example/NORMAL_SNP.formatted.txt
=================================================
Arguments are:
Query File: example/Example.intervals
k: 3
tau: 2
Output Directory: ./
Output Prefix: Example
Num Processes: 5
Graph extension: .pdf
Valid sample for THetA analysis:
Ratio Deviation: 0.1
Min Fraction of Genome Aberrated: 0.05
Program WILL cluster intervals.
=================================================
Reading in query file...
[...snip...]
[user@cn3144 ~]$ ls -lh
total 232K
drwxr-xr-x 2 user group 4.0K Oct 13 10:32 example
drwxr-xr-x 9 user group 4.0K Oct 13 16:19 Example_2_cluster_data
drwxr-xr-x 8 user group 4.0K Oct 13 16:20 Example_3_cluster_data
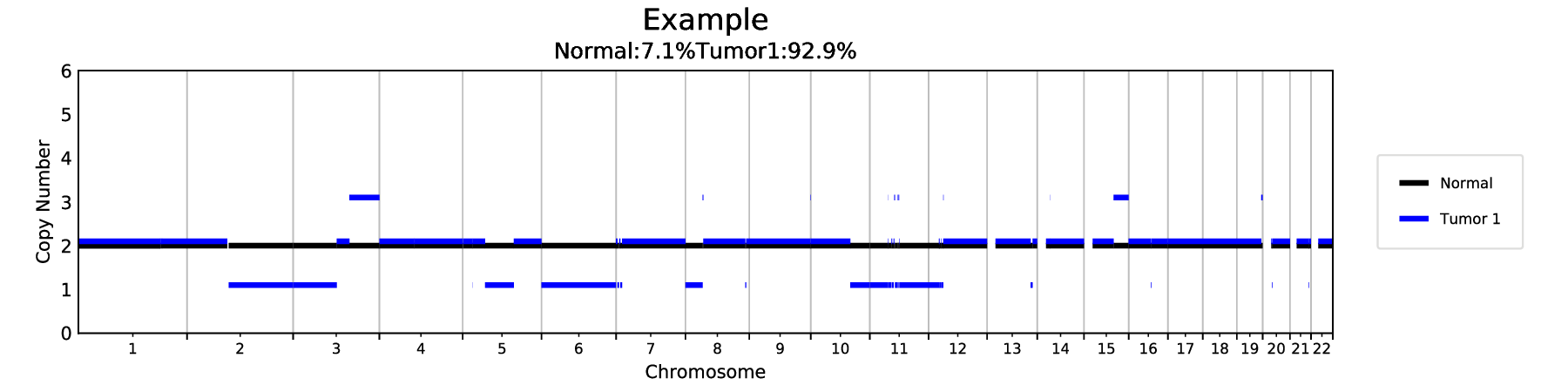
-rw-r--r-- 1 user group 13K Oct 13 16:19 Example_assignment.png
-rw-r--r-- 1 user group 2.0K Oct 13 16:20 Example.BEST.results
-rw-r--r-- 1 user group 118K Oct 13 16:19 Example_by_chromosome.png
-rw-r--r-- 1 user group 17K Oct 13 16:19 Example_classifications.png
-rw-r--r-- 1 user group 16K Oct 13 16:19 Example.n2.graph.pdf
-rw-r--r-- 1 user group 2.0K Oct 13 16:19 Example.n2.results
-rw-r--r-- 1 user group 3.6K Oct 13 16:19 Example.n2.withBounds
-rw-r--r-- 1 user group 17K Oct 13 16:20 Example.n3.graph.pdf
-rw-r--r-- 1 user group 2.2K Oct 13 16:20 Example.n3.results
-rw-r--r-- 1 user group 3.6K Oct 13 16:20 Example.n3.withBounds
-rw-r--r-- 1 user group 225 Oct 13 16:19 Example.RunN3.bash
The analysis will create a number of files including some graphs. For example, the following shows one of the models (2 components):
In addition to RunTHetA there are several other tools included
in this package
[user@cn3144 ~]$ ls /usr/local/apps/theta/0.7-20-g94fd772/bin |-- CreateExomeInput |-- getAlleleCounts |-- runBICSeqToTHetA `-- RunTHetA
2 of these tools (getAlleleCounts and runBICSeqToTHetA) are wrappers around
java tools. In addition to their normal arguments they also take the --java-opts
argument which can be used to pass options to java
[user@cn3144 ~]$ runBICSeqToTHetA --java-opts="-Xmx2g" --help
Error! Incorrect number of arguments.
Program: BICSeqToTHetA
USAGE (src): java BICSeqToTHetA <INPUT_FILE> [Options]
USAGE (jar): java -jar BICSeqToTHetA <INPUT_FILE> [Options]
<INPUT_FILE> [String]
A file output by BIC-Seq.
-OUTPUT_PREFIX [STRING]
Prefix for all output files.
-MIN_LENGTH [Integer]
The minimum length of intervals to keep.
For a more detailed manual see
/usr/local/apps/theta/<version>/MANUAL.txt
[user@cn3144 ~]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf ~]$
Create a batch input file (e.g. THetA.sh), which uses the input file 'THetA.in'. For example:
#! /bin/bash module load theta/0.7-20-g94fd772 || exit 1 RunTHetA example/Example.intervals \ --NUM_PROCESSES=$((SLURM_CPUS_PER_TASK - 1)) \ --TUMOR_FILE example/TUMOR_SNP.formatted.txt \ --NORMAL_FILE example/NORMAL_SNP.formatted.txt
Submit this job using the Slurm sbatch command.
sbatch --cpus-per-task=6 --mem=14g theta.sh
Create a swarmfile (e.g. THetA.swarm). For example:
RunTHetA sample1/Example.intervals --NUM_PROCESSES=$((SLURM_CPUS_PER_TASK-1)) \ --TUMOR_FILE sample1/TUMOR_SNP.formatted.txt --NORMAL_FILE sample2/NORMAL_SNP.formatted.txt RunTHetA sample2/Example.intervals --NUM_PROCESSES=$((SLURM_CPUS_PER_TASK-1)) \ --TUMOR_FILE sample2/TUMOR_SNP.formatted.txt --NORMAL_FILE sample2/NORMAL_SNP.formatted.txt
Submit this job using the swarm command.
swarm -f THetA.swarm -g 14 -t 6 --module thetawhere
| -g # | Number of Gigabytes of memory required for each process (1 line in the swarm command file) |
| -t # | Number of threads/CPUs required for each process (1 line in the swarm command file). |
| --module THetA | Loads the THetA module for each subjob in the swarm |