CHARMM (Chemistry at HARvard Macromolecular Mechanics) [1]:
As of October, 2020, CHARMM version c45b1 has been made available on Biowulf for general use. There are important changes in how to run this version. In particular the "cover scripts" that were previously used to run CHARMM have been retired. Instead, users must explicitly run mpirun or srun for parallel runs and call the charmm program directly for serial runs.
For documentation on running CHARMM c42b2 and below with cover scripts, please see the documentation for older releases.
The following modules are available:
For a non-parallel CHARMM job such as model building or ad hoc trajectory analysis, the commands and setup have few requirements. The job script (build.csh) can be simply:
#!/bin/csh cd $SLURM_SUBMIT_DIR module load charmm/c45b1/serial charmm < build-psf.inp >& build-psf.out
The above can be submitted to the batch queue via:
sbatch build.csh
For parallel usage, the following script (sbatch.sh) illustrates submitting a SLURM batch job which will use the 28 physical cores on each of 4 nodes (112 total cores:
#!/bin/bash
#SBATCH --partition=multinode
#SBATCH --time=04:00:00
#SBATCH --ntasks=112
#SBATCH --nodes=4
#SBATCH --ntasks-per-core=1
#SBATCH --constraint="[x2680|x2695]"
module load charmm/c45b1/domdec
srun --mpi=pmix_v3 --ntasks=${SLURM_NTASKS} charmm -i input.inp -o output.out
The environment variable SLURM_SUBMIT_DIR points to the working directory where 'sbatch' was run, and SLURM_NTASKS contains the value given with the --ntasks= argument to sbatch. The above is suitable for most parallel CHARMM usage, other than the DOMDEC_GPU code (see below).
Note that it is possible to specify sbatch parameters on the command line rather than in the batch script itself. Command line parameters override those specified within the batch script. E.g. sbatch --ntasks=56 --nodes=2 --ntasks-per-core=1 --partition=multinode sbatch.sh would submit the job to 2 nodes via the command line.
The DOMDEC_GPU code may be used by using the charmm/c45b1/gpu module. DOMDEC_GPU uses both an MPI library (OpenMPI in this case) and OpenMP threads, and therefore requires changes to the SLURM sbatch arguments. The changes are shown in the example below (sbatchGPU.csh);
#!/bin/bash #SBATCH --partition=gpu #SBATCH --ntasks=1 #SBATCH --cpus-per-task=8 #SBATCH --gres=gpu:k80:1 #SBATCH --time=4:00:00 export OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK module load charmm/c45b1/gpu srun --mpi=pmix_v3 --ntasks=$SLURM_NTASKS --cpus-per-task=$SLURM_CPUS_PER_TASK charmm -i input.inp -o output.out
This script runs DOMDEC_GPU on a single Biowulf K80 GPU and can be submitted with sbatch sbatchGPU.sh. As with the CPU script, arguments can be given on the command line rather than in the script. Remember to modify the wall time as needed!
For a variety of tasks such as model building, analysis, and graphics, foreground interactive use of CHARMM can be advantageous, esp. when developing and testing a new input script. The SLURM sinteractive command makes this fairly easy (system prompts in bold, user input in italics):
biowulf /<2>EwaldNVE [69] sinteractive salloc.exe: Granted job allocation 1693180 salloc.exe: Waiting for resource configuration salloc.exe: Nodes cn0032 are ready for job cn0032 /<2>EwaldNVE [1] module load charmm/c45b1/serial cn0032 /<2>EwaldNVE [2] charmm < build-psf.inp >& build-psf.out cn0032 /<2>EwaldNVE [3] exit exit salloc.exe: Relinquishing job allocation 1693180 salloc.exe: Job allocation 1693180 has been revoked.
For troubleshooting, it may be useful to pipe the output, and both save it in file (via 'tee') and view it in the 'less' browser, e.g.:
cn0254 /<2>EwaldNVE [3] charmm < minmodel.inp |& tee minmodel.out | less
Finally, CHARMM itself can be run interactively, via simply:
biowulf /<2>EwaldNVE [71] sinteractive
salloc.exe: Granted job allocation 1693592
salloc.exe: Waiting for resource configuration
salloc.exe: Nodes cn0103 are ready for job
cn0103 /<2>EwaldNVE [1] module load charmm/c45b1/serial
cn0103 /<2>EwaldNVE [2] charmm
Chemistry at HARvard Macromolecular Mechanics
(CHARMM) - Developmental Version 45b1 August 15, 2020
Revision unknown
Copyright(c) 1984-2020 President and Fellows of Harvard College
All Rights Reserved
Current operating system: Linux-3.10.0-862.14.4.el7.x86_64(x86_64)@cn0858
Created on 10/15/20 at 11:58:24 by user: btmiller
Maximum number of ATOMS: 360720, and RESidues: 120240
At this point the program is expecting input, starting with a title; it is recommended to type bomlev -1 as the first command, as that will forgive typing errors and allow the program to continue. It also recommended to have the initial setup commands (reading RTF and PARAM files, PSF and COOR files, etc.) in a 'stream' file, so that those actions can be done vie e.g.
stream init.str
The same applies to other complex setups, such as establishing restraints, or graphics setup.
Note that the graphics uses X11, so the initial login to biowulf should use either the -X or the -Y option of the ssh command to enable X11 tunneling or use Open OnDemand for the graphics display.
Recent versions of the distributed CHARMM parameters, including the latest release, are available in /usr/local/apps/charmm as subdirectories topparYYYY where YYYY is the release year. Each release contains a number of corrections and additions from the past year, esp. for the CHARMM force fields. The toppar2017 and later releases uses a newer format, so one should be careful about using it with older PSF files. Files distributed with models built using CHARMM-GUI now use this newer format.
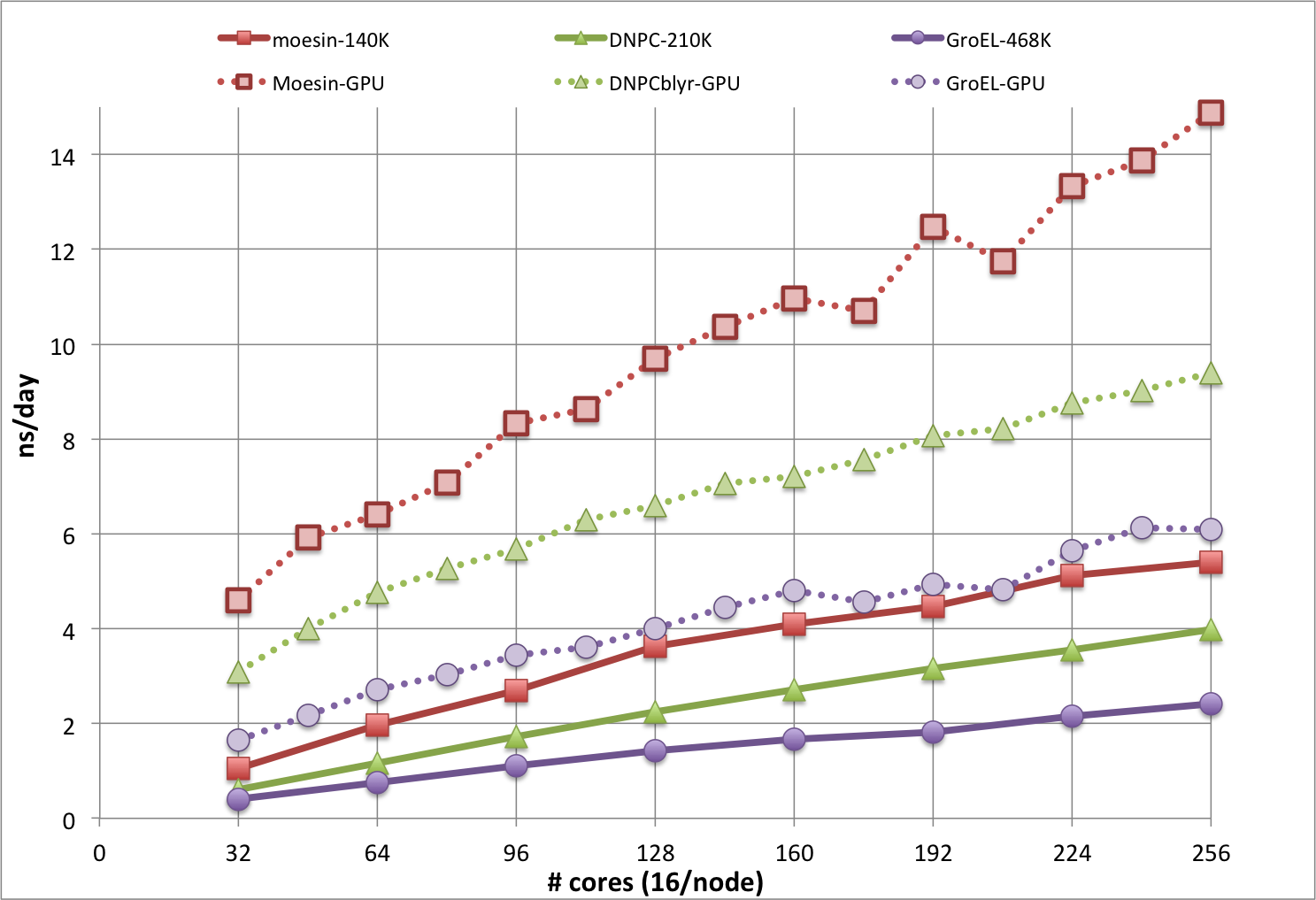
The plot below is from CHARMM benchmarks run during the beta test phase of Biowulf, and shows results for DOMDEC on Infiniband nodes (solid lines) and DOMDEC_GPU on gpu nodes (dotted lines), for even numbers from 2 through 16 nodes. The timings in ns/day are from short MD simulations, run with a 1 fs integration time step, for 3 molecular systems of different sizes and shapes:
Note that the ns/day rate would be doubled with the use of a 2 fs time step, which is often done for more exploratory sampling, but not necessarily recommended for the best accuracy and precision. Simulations systems that cannot use DOMDEC will be somewhat slower, and will not scale well past about 64 cores.