The MPSAN_MP application employs neural networks implementing the Message Passing or Self-Attention
mechanism for Molecular Property prediction. It takes as input a set of molecules represented by
either graphs or SMILES strings. For each molecule, it predicts a propery value (PV), which may be either
a discrete/binary label (classification task) or a continuous value (regression task).
of molecular property prediction:
This application is been be used as a biological example by the class #7 of the course
"Deep Learning by Example on Biowulf".
Allocate an interactive session and run the program. Sample session:
[user@biowulf]$ sinteractive --mem=64g --gres=gpu:a100:1,lscratch:100 -c24 [user@cn2893 ~]$module load mpsan_mp [+] Loading singularity 4.0.1 on cn2893 [+] Loading mpsan_mp 20240817(a lower-case name, mpsan_mp, will also work).
[user@cn2893 ~]$ preprocess.py -h
usage: preprocess.py [-h] [-a] [-A augm_coeff] [-b] -d data_name -i input_file -o output_file [-r]
optional arguments:
-h, --help show this help message and exit
-a, --augment augment the data by enumerating SMILES
-A augm_coeff, --augm_coeff augm_coeff
augmentation_coefficient; fault = 10
-b, --binarize binarize the logp data
-r, --reformat reformat the data to make them acceptable by MPTN_MP software
required arguments:
-d data_name, --data_name data_name
Data type: jak2 | logp | bbbp | chembl | zinc
-i input_file, --input_file input_file
input file; fault = None
-o output_file, --output_file output_file
output file
The executable expects that the input file has been downladed and/or is available in the csv format. [user@cn2893 ~]$ wget https://ftp.ebi.ac.uk/pub/databases/chembl/ChEMBLdb/releases/chembl_33/chembl_33_chemreps.txt.gz [user@cn2893 ~]$ gunzip chembl_33_chemreps.txt.gz [user@cn2893 ~]$ preprocess.py -r -d chembl -i chembl_33_chemreps.txt -o chembl_natoms,ntokens.csv- to augment jak2 data:
[user@cn2893 ~]$cp $MPSAN_MP_DATA/jak2_natoms,ntokens.csv . [user@cn2893 ~]$preprocess.py -a -d jak2 -i jak2_natoms,ntokens.csv -o jak2a_natoms,ntokens.csv- to binarize the logp data:
[user@cn2893 ~]$cp $MPSAN_MP_DAT/logp_natoms,ntokens.csv . [user@cn2893 ~]$preprocess.py -b -d logp -i logp_natoms,ntokens.csv -o blogp_natoms,ntokens.csvBefore using other executables, create a local folder "data" and copy one or more original data files into that folder:
[user@cn2893 ~]$ mkdir -p data [user@cn2893 ~]$ cp $MPSAN_MP_DATA/* dataThe mode of usage of the executable train.py depends on the (required) data and model names passed to it through command line options:
[user@cn2893 ~]$ train.py -h
Using TensorFlow backend
usage: train.py [-h] [-A num_heads] [-B num_ln_layers] [-D dropout_rate] [--data_path data_path]
[--debug DEBUG] [--embed_dim EMBED_DIM] [--frac_valid FRAC_VALID] [--frac_test FRAC_TEST]
[-l learning_rate] [-i input_checkpoint_file] [-I encoder_checkpoint_file] [--LF L]
[-E num_encoder_layers] [--num_ft_layers num_ft_layers] [--num_t_layers num_t_layers] -m
model_name [-M M] [--max_length MAX_LENGTH] [--mp_iterations mp_iterations] [--NF N]
[-e num_epochs] [-O optimizer] [-p pdata_nane] [-P POSITION_EMBEDDING] [-T] [-t TOKENIZER]
[--trainable_encoder] [-U num_dense_units] [-v] [-w] [-b batch_size]
[--best_accuracy best_accuracy] [--best_loss best_loss] -d DATA_NAME [-n NUM_DATA]
[--es_patience patience] [-S smiles]
optional arguments:
-h, --help show this help message and exit
-A num_heads, --num_heads num_heads
number of attention heads; default=4
-B num_ln_layers, --num_ln_layers num_ln_layers
number of LayerNormalization layers; max = 2; default=2
-D dropout_rate, --dropout_rate dropout_rate
dropout_rate; default = 0.1
--data_path data_path
path to data file (overrides the option -d
--debug DEBUG use a small subset of data for training
--embed_dim EMBED_DIM
the dimension of the embedding space; default: 1024
--frac_valid FRAC_VALID
fraction of data to be used for validation; default=0.05
--frac_test FRAC_TEST
fraction of data to be used for testing; default=0.15
-l learning_rate, --lr learning_rate
learning rate; default=1.e-4
-i input_checkpoint_file, --input_checkpoint_file input_checkpoint_file
input checkpoint file; fault = None
-I encoder_checkpoint_file, --input_encoder checkpoint_file encoder_checkpoint_file
input encoder checkpoint file; fault = None
--LF L num of link/edge features in synthetic data; default=2
-E num_encoder_layers, --num_enc_layers num_encoder_layers
number of layers in BERT Encoder; default=6
--num_ft_layers num_ft_layers
number of dense layers in finetuner: 1, 2 or 3; default=1
--num_t_layers num_t_layers
number of transformer layers in the san model; default=1
-M M num of nodes/atoms in synthetic data; default=5
--max_length MAX_LENGTH
max # tokens of input SMILES to be used for training; default=64
--mp_iterations mp_iterations
# of MP iterations; default=4
--NF N num of node/atom features in synthetic data; default=3
-e num_epochs, --num_epochs num_epochs
# of epochs; default=40 if data_name=="chembl", =3000 otherwise
-O optimizer, --optimizer optimizer
optimizer: adam | rmsprop
-p pdata_nane, --pretraining_data_name pdata_nane
name of the data trhat were used at pretraining step
-P POSITION_EMBEDDING, --position_embedding POSITION_EMBEDDING
0=none(default);1=regular,2=sine;3=regular_concat;4=sine_concat
-T, --transpose transpose/permute data tensor after embedding
-t TOKENIZER, --tokenizer TOKENIZER
tokenizer used by san-based model: smiles_pe or atom_symbols; default=smiles_pe
--trainable_encoder Encoder is trainable even if not data=chembl; default=False
-U num_dense_units, --num_dense_units num_dense_units
number of dense units; default=1024
-v, --verbose increase the verbosity level of output
-w, --load_weights read weights from a checkpoint file
-b batch_size, --bs batch_size
batch size; default=256
--best_accuracy best_accuracy
initial value of best_accuracy; default = 0
--best_loss best_loss
initial value of best_loss; default = 10000
-n NUM_DATA, --num_data NUM_DATA
number of input SMILES strings to be used; default=all
--es_patience patience
patience for early stopping of training; default=0 (no early stopping)
-S smiles, --smiles smiles
(Comma-separated list of) smiles to be used as input for debugging
required arguments:
-m model_name, --model_name model_name
model name: mpn | san | san_bert; default = mpn
required arguments:
-d DATA_NAME, --data_name DATA_NAME
Data type: jak2(a) | (b)logp(a) | bbbp(a) | chembl | zinc
Examples of usage of the train.py executable: [user@cn2893 ~]$ train.py -m mpn -d jak2 # training mode- a similar command to train san model on augmented logp data:
[user@cn2893 ~]$ train.py -m san -d logpa # training mode ...- to pretrain san_bert model on chembl data:
[user@cn2893 ~]$train.py -m san_bert -d chembl # pretraining mode- to finetune the san_bert on bbbp data, using the source model pretrained on zinc data:
[user@cn2893 ~]$train.py -d bbbp -m san_bert -p zinc \ # finetuning mode
-i checkpoints/san_bert.zinc.bbbpa.weights.h5 \
-I checkpoints/bert_encoder.zinc.san_bert.weights.h5
...
Correct= 243 / 268 %error= 9.328358208955223
- a similar command, but using chembl as the source dataset:
[user@cn2893 ~]$train.py -d bbbp -m san_bert -p chembl \ # finetuning mode
-i checkpoints/san_bert.chembl.bbbpa.weights.h5 \
-I checkpoints/bert_encoder.chembl.san_bert.weights.h5
...
Correct= 251 / 268 %error= 6.343283582089554
The command line options for the executable predict.py are similar to, and should be consistent with,
the options that were passed to the training command:
[user@cn2893 ~]$ predict.py -h
Using TensorFlow backend
usage: predict.py [-h] [-A num_heads] [-B num_ln_layers] [-D dropout_rate] [--data_path data_path]
[--debug DEBUG] [--embed_dim EMBED_DIM] [--frac_valid FRAC_VALID] [--frac_test FRAC_TEST]
[-l learning_rate] [-i input_checkpoint_file] [-I encoder_checkpoint_file] [--LF L]
[-E num_encoder_layers] [--num_ft_layers num_ft_layers] [--num_t_layers num_t_layers] -m
model_name [-M M] [--max_length MAX_LENGTH] [--mp_iterations mp_iterations] [--NF N]
[-e num_epochs] [-O optimizer] [-p pdata_nane] [-P POSITION_EMBEDDING] [-T] [-t TOKENIZER]
[--trainable_encoder] [-U num_dense_units] [-v] [-w] [-b batch_size] -d DATA_NAME
optional arguments:
-h, --help show this help message and exit
-A num_heads, --num_heads num_heads
number of attention heads; default=4
-B num_ln_layers, --num_ln_layers num_ln_layers
number of LayerNormalization layers; max = 2; default=2
-D dropout_rate, --dropout_rate dropout_rate
dropout_rate; default = 0.1
--data_path data_path
path to data file (overrides the option -d
--debug DEBUG use a small subset of data for training
--embed_dim EMBED_DIM
the dimension of the embedding space; default: 1024
--frac_valid FRAC_VALID
fraction of data to be used for validation; default=0.05
--frac_test FRAC_TEST
fraction of data to be used for testing; default=0.15
-l learning_rate, --lr learning_rate
learning rate; default=1.e-4
-i input_checkpoint_file, --input_checkpoint_file input_checkpoint_file
input checkpoint file; fault = None
-I encoder_checkpoint_file, --input_encoder checkpoint_file encoder_checkpoint_file
input encoder checkpoint file; fault = None
--LF L num of link/edge features in synthetic data; default=2
-E num_encoder_layers, --num_enc_layers num_encoder_layers
number of layers in BERT Encoder; default=6
--num_ft_layers num_ft_layers
number of dense layers in finetuner: 1, 2 or 3; default=1
--num_t_layers num_t_layers
number of transformer layers in the san model; default=1
-M M num of nodes/atoms in synthetic data; default=5
--max_length MAX_LENGTH
max # tokens of input SMILES to be used for training; default=64
--mp_iterations mp_iterations
# of MP iterations; default=4
--NF N num of node/atom features in synthetic data; default=3
-e num_epochs, --num_epochs num_epochs
# of epochs; default=40 if data_name=="chembl", =3000 otherwise
-O optimizer, --optimizer optimizer
optimizer: adam | rmsprop
-p pdata_nane, --pretraining_data_name pdata_nane
name of the data trhat were used at pretraining step
-P POSITION_EMBEDDING, --position_embedding POSITION_EMBEDDING
0=none(default);1=regular,2=sine;3=regular_concat;4=sine_concat
-T, --transpose transpose/permute data tensor after embedding
-t TOKENIZER, --tokenizer TOKENIZER
tokenizer used by san-based model: smiles_pe or atom_symbols; default=smiles_pe
--trainable_encoder Encoder is trainable even if not data=chembl; default=False
-U num_dense_units, --num_dense_units num_dense_units
number of dense units; default=1024
-v, --verbose increase the verbosity level of output
-w, --load_weights read weights from a checkpoint file
-b batch_size, --bs batch_size
batch size; default=256
required arguments:
-m model_name, --model_name model_name
model name: mpn | san | san_bert; default = mpn
required arguments:
-d DATA_NAME, --data_name DATA_NAME
Data type: jak2(a) | (b)logp(a) | bbbp(a) | chembl | zinc
For example, [user@cn2893 ~]$ predict.py -m mpn -d jak2 -i checkpoints/mpn.jak2.weights.h5 ...the results will be output to the file results/mpn.jak2.results.tsv:
[user@cn2893 ~]$ cat results/mpn.jak2.results.tsv # pred_jak2 corr_jak2 5.705802 7.080000 7.524415 6.460000 8.072280 7.050000 7.216453 8.220000 8.825060 9.260000 6.521371 7.600000 6.853940 6.820000 8.652765 8.050000 6.496820 7.460000 ... 7.646416 7.600000 7.397765 7.820000 7.131140 7.270000 6.656110 8.150000 7.219415 7.600000Finally, in order to visualize the summary provided in the results file, run the executable visualize.R. To display the usage message of this executable, type its name:
[user@cn2893 ~]$ module load R [user@cn2893 ~]$ visualize.R Usage: visualize.R <results_file> [ <other_results_file(s)> ]Examples of usage of this executable:
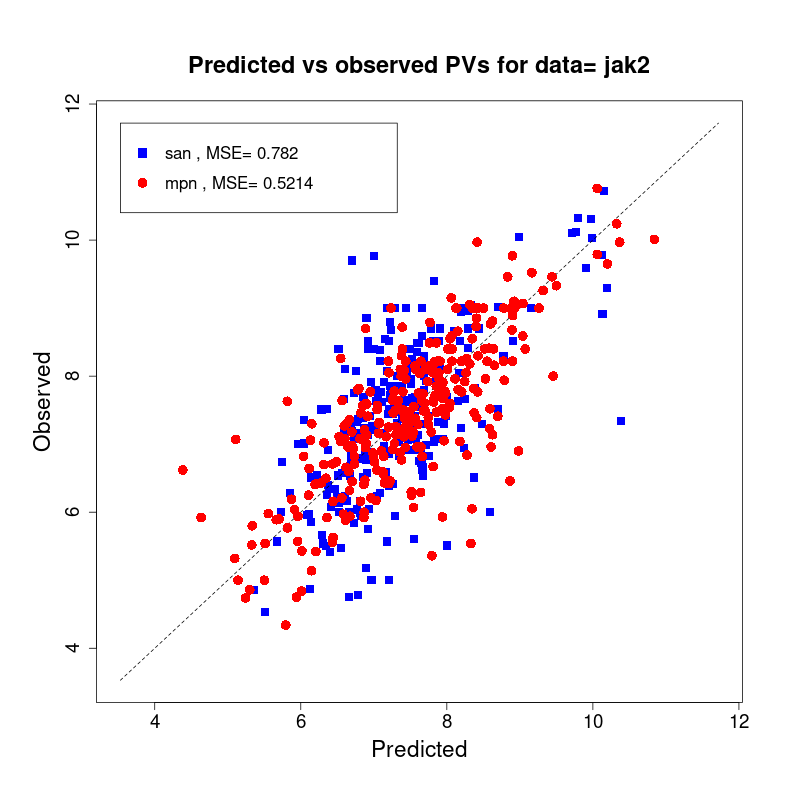
[user@cn2893 ~]$ visualize.R results/san.jak2.results.tsv results/mpn.jak2.results.tsv
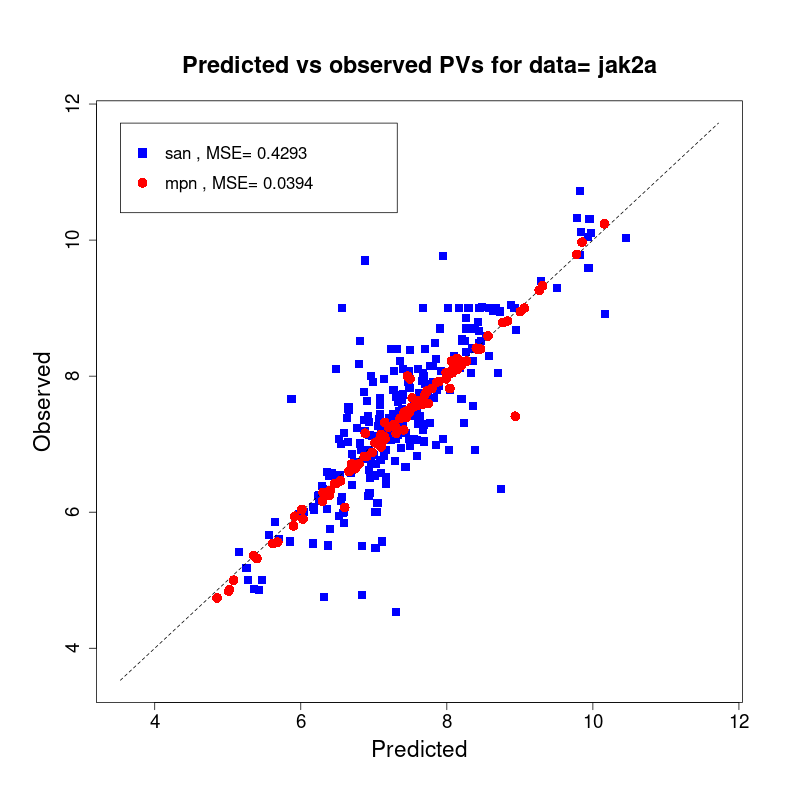
[user@cn2893 ~]$ visualize.R results/san.jak2a.results.tsv results/mpn.jak2a.results.tsv
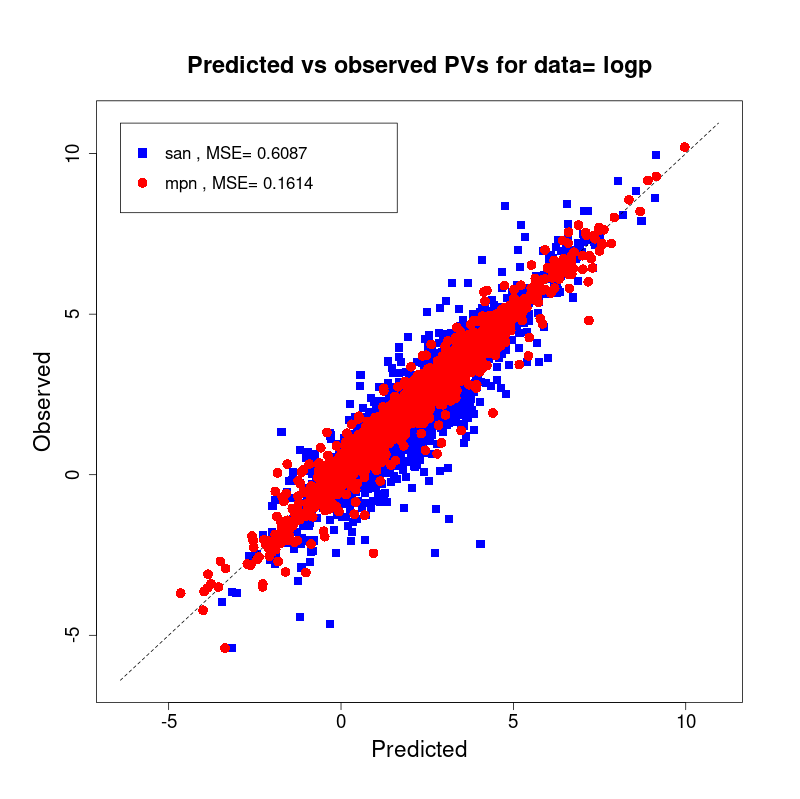
[user@cn2893 ~]$ visualize.R results/san.logp.results.tsv results/mpn.logp.results.tsv
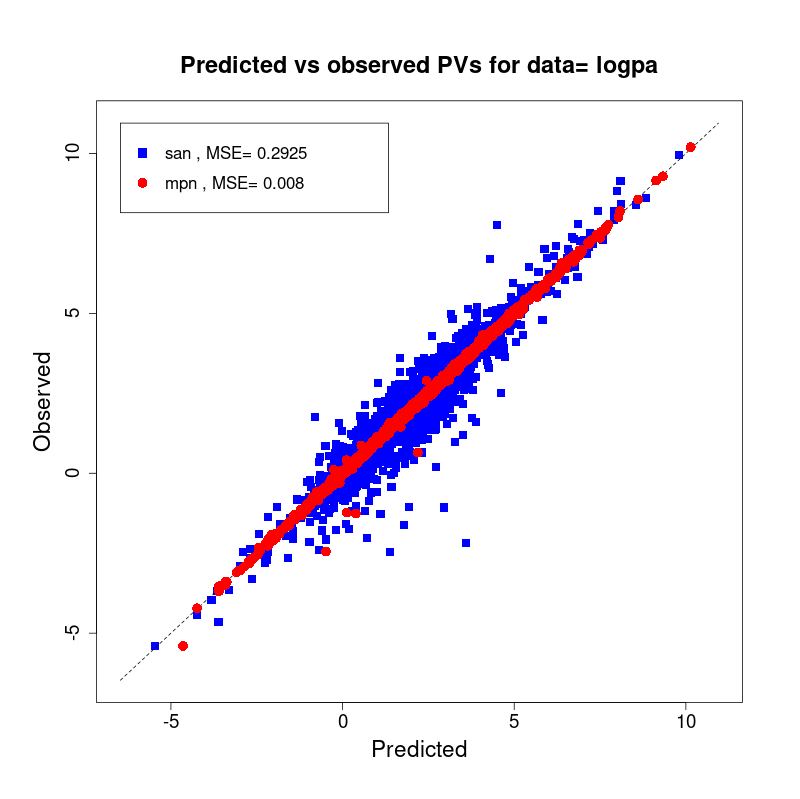
[user@cn2893 ~]$ visualize.R results/san.logpa.results.tsv results/mpn.logpa.results.tsv
[user@cn2893 ~]$ exit salloc.exe: Relinquishing job allocation 46116226 [user@biowulf ~]$