Implicit Solvent
January 2021
The benchmark runs below used the Amber 20 Benchmark suite, downloadable from here.
Older benchmarks: [Amber 18] Amber 16] [Amber 14]
Hardware:
Amber 20 with all patches as of Nov 2020.
CPU runs: Amber 20 built with Intel 2020.0.166 compilers, CUDA 10.2, OpenMPI 4.0.4 (Biowulf module 'amber/20.intel')
GPU runs: Amber 20 built with gcc 7.4, CUDA 10.1, and OpenMPI 4.0.4 (Biowulf module 'amber/20-gpu')
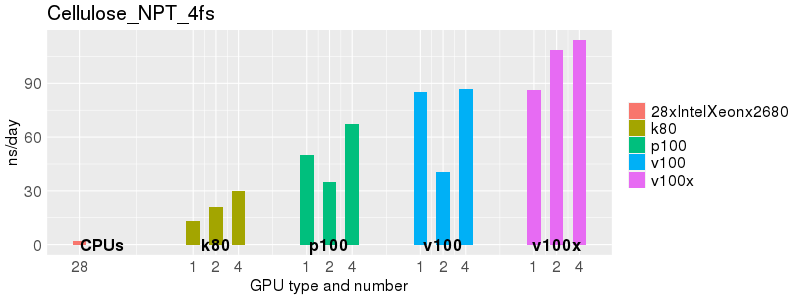
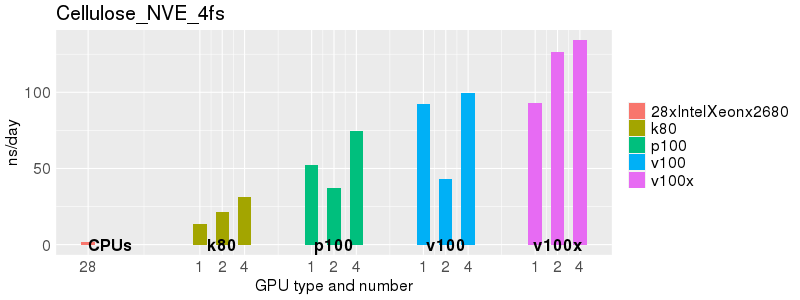
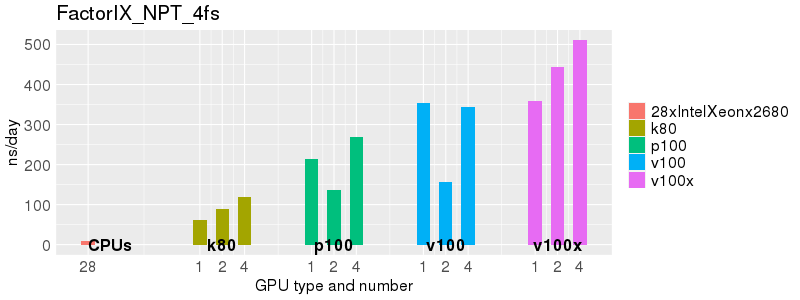
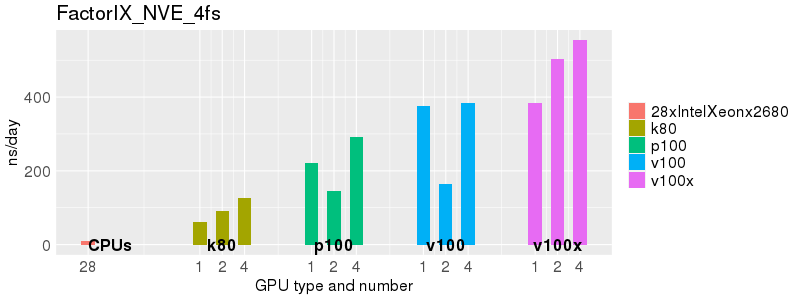
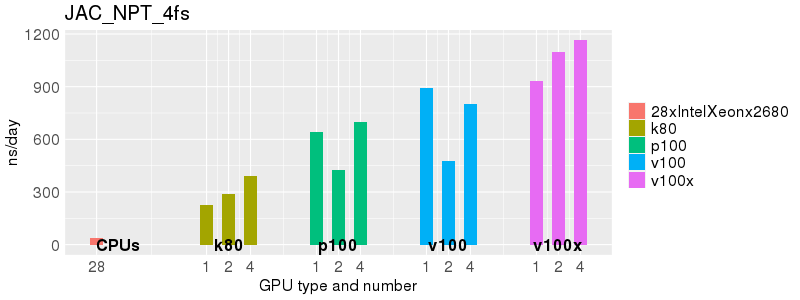
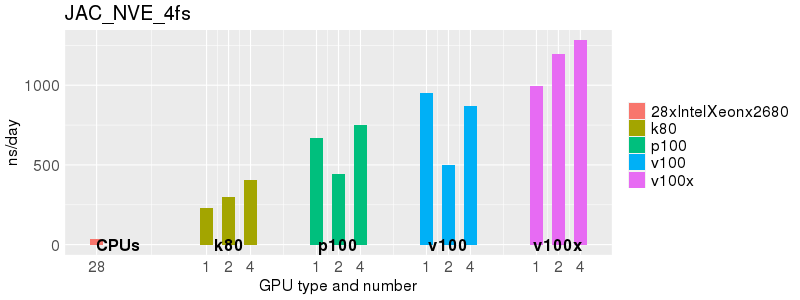
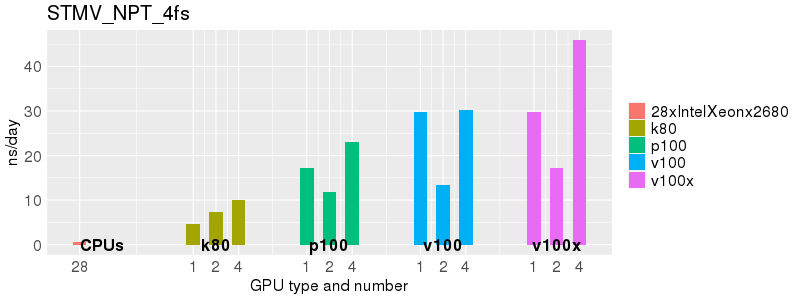
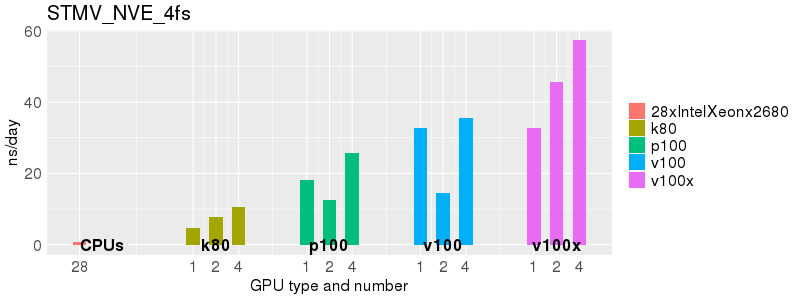
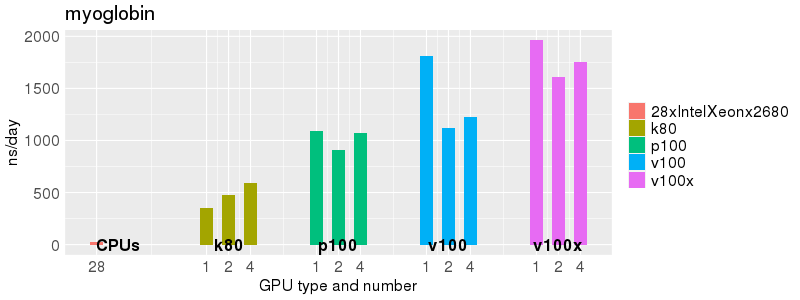
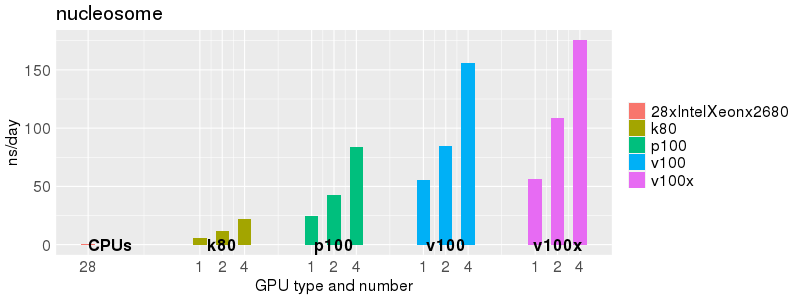
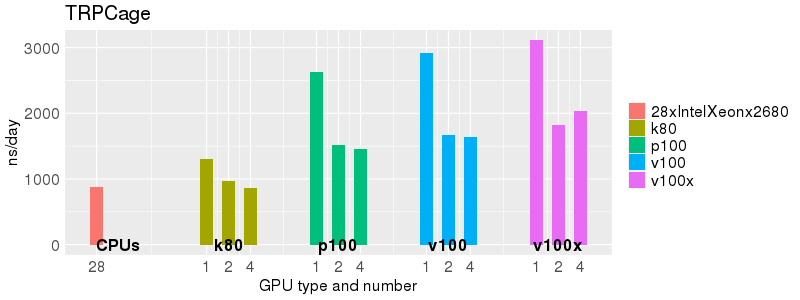
Based on these benchmarks, there is a significant performance advantage to running Amber on the GPU nodes, especially the P100s and V100s, rather than on a CPU-only node.
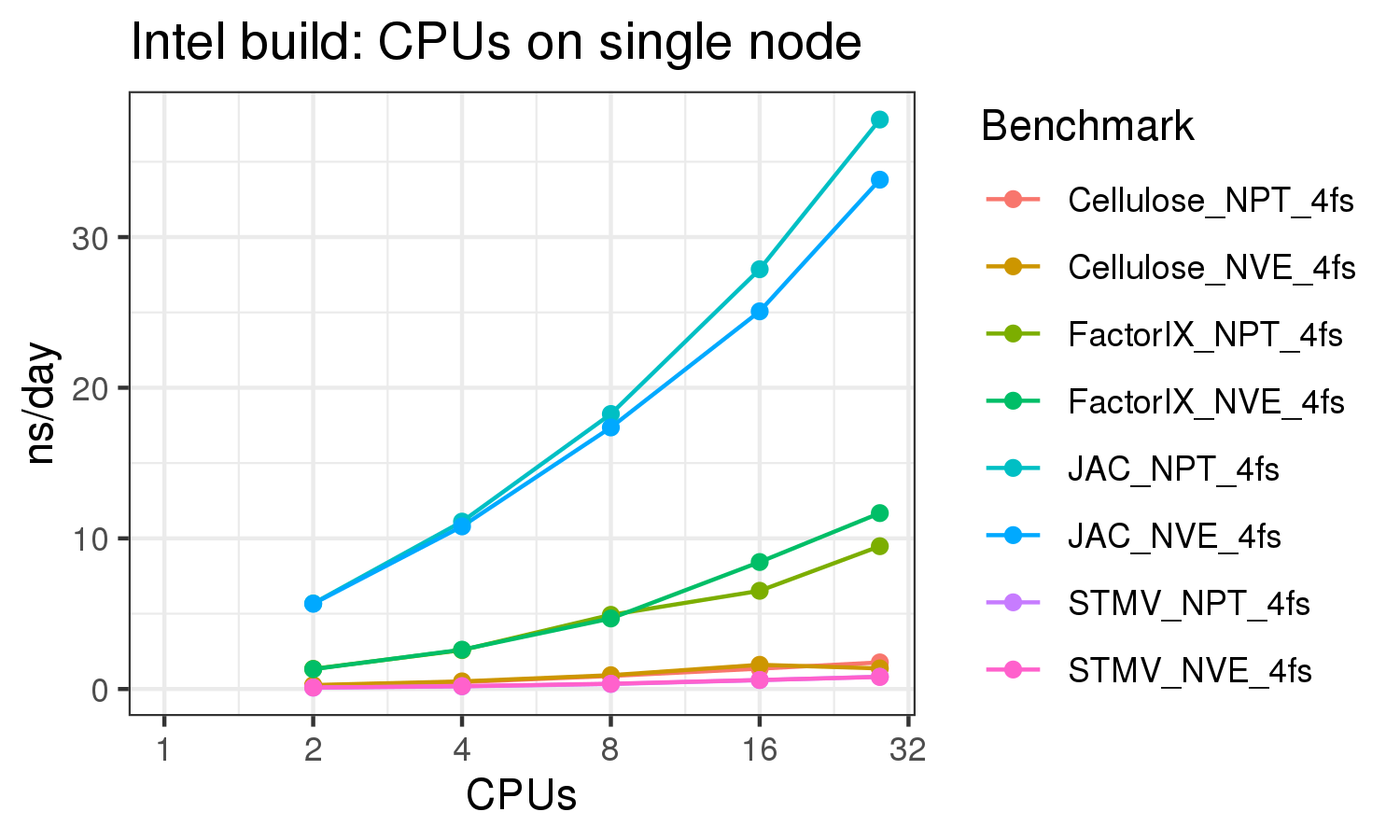
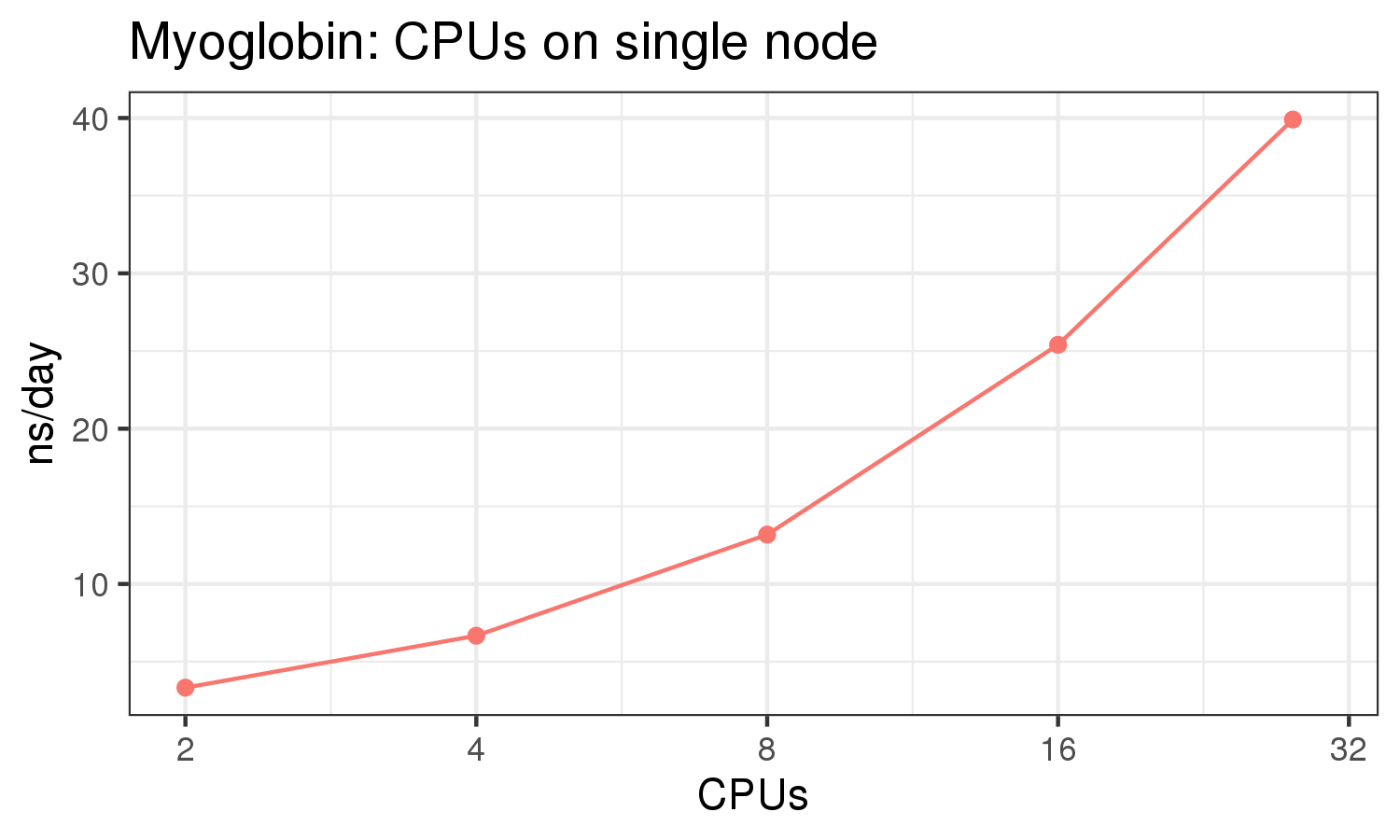
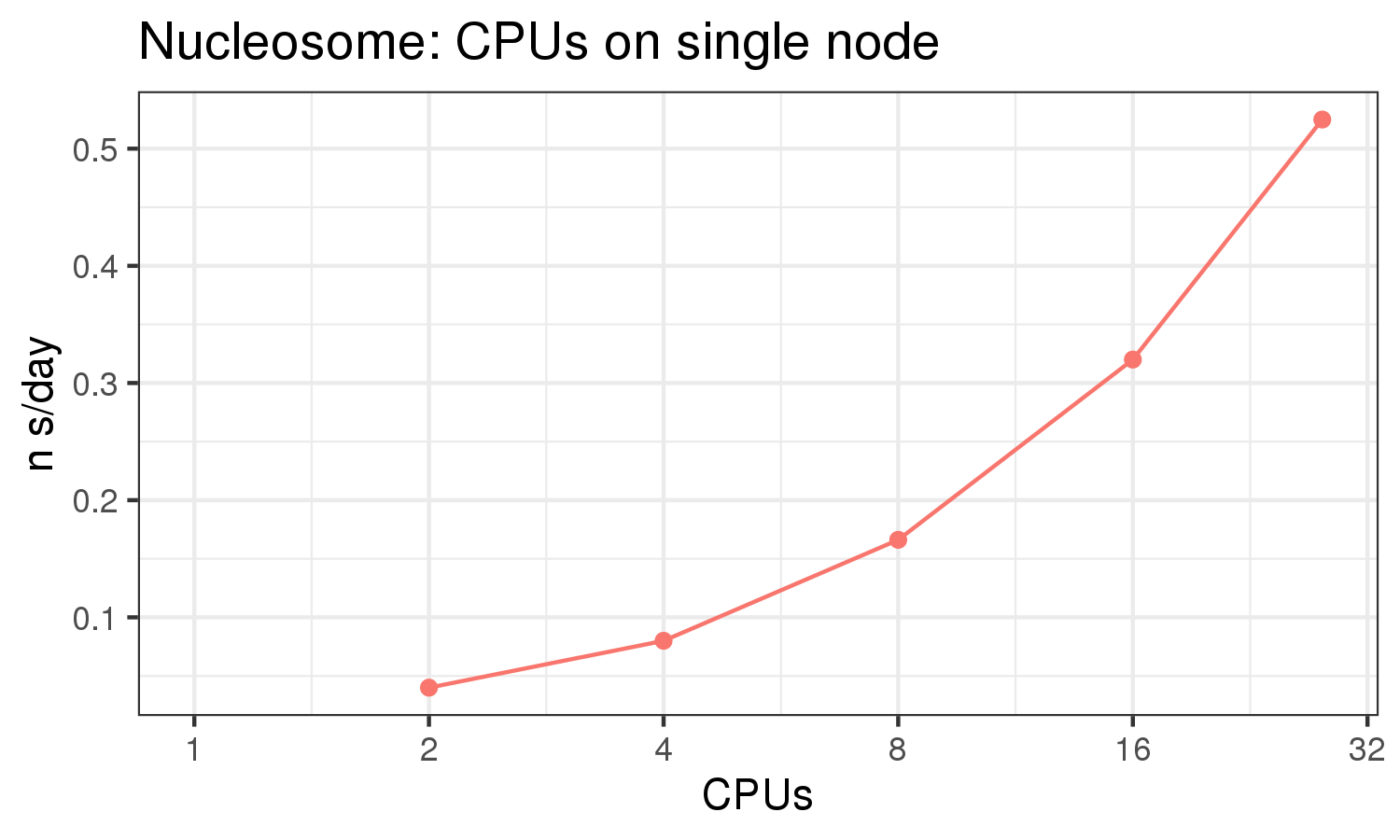
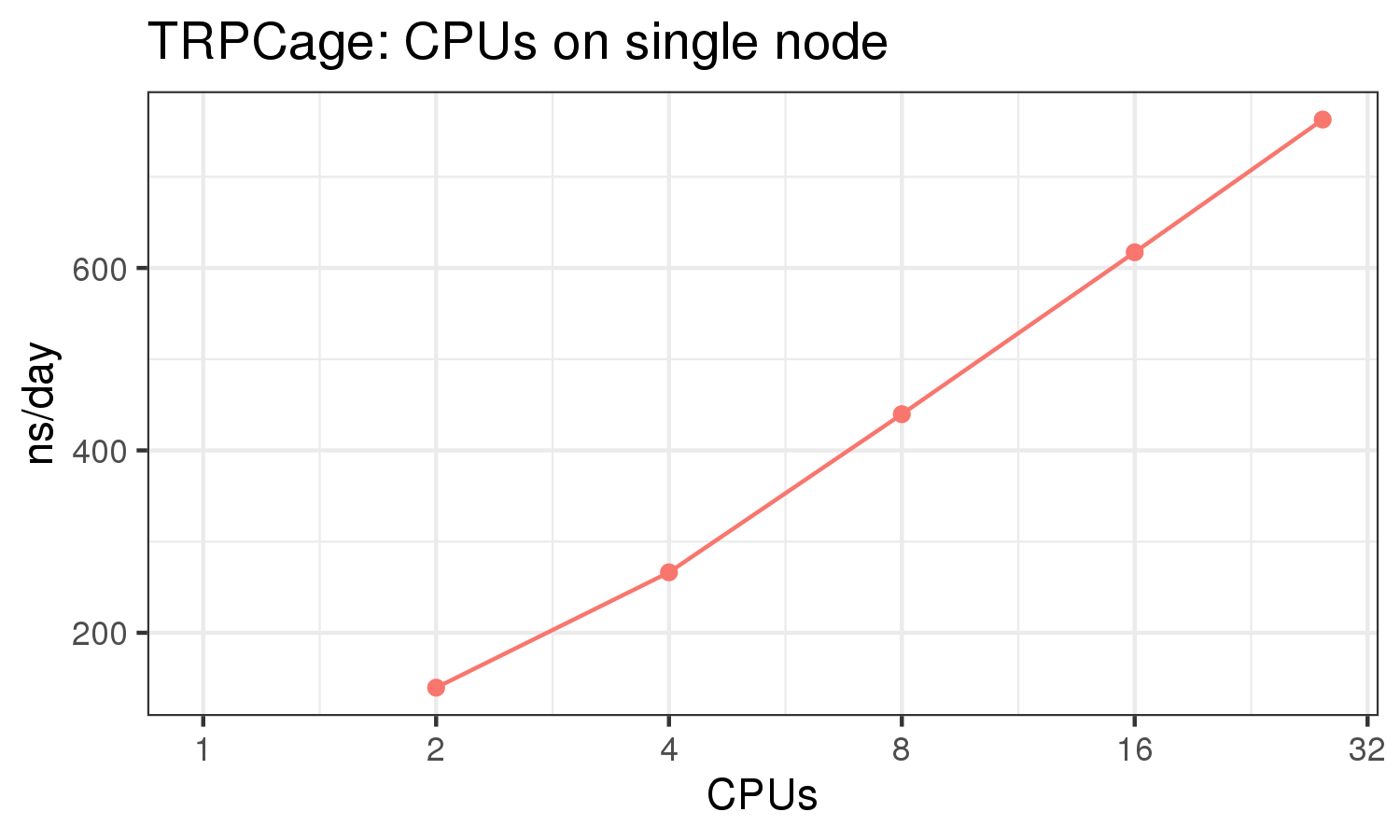
The benchmark above was run on a Intel Xeon E5-2680 v4 @2.5GHz with 28 cores (56 hyperthreaded cores). As with other MD applications, the performance drops when more than 28 MPI processes (i.e. 1 process per physical core) are run. Thus, if running on CPUs, it is important to use the '--ntasks-per-core=1' flag when submitting the job, to ensure that only 1 MPI process is run on each physical core. If it is possible to run on a GPU node, you will get significantly better performance as shown in the benchmarks below.
Implicit Solvent (GB)
Explicit Solvent (PME)